
La sicklemia es una enfermedad genética que puede pasar inadvertida por las personas que la padecen.
Y puede ser el caso de cualquier persona que no conozca que es portador del gen causal de esta enfermedad y que por lo tanto puede transmitirla a su descendencia.
Y esto se detecta a traves de las pruebas prenatales que habitualmente se realizan durante el embarazo las que revelan su condición. Puede presentarse sin haber experimentado alguna molestia relacionada con la sicklemia, y a punto de partida de este diagnostico se deben tomar algunas medidas para no poner en riesgo la salud de la persona.
Y esto puede suceder en personas con rasgos europeos, pero que en su ascendencia se encuentran antecedentes de personas de origen africano, lo que puede dar lugar a su estatus de portador del gen de la sicklemia.
Más artículos de enfermedades: EL ALZHEIMER, ESE GRAN OLVIDADO
Y en estas circunstancias el conyuge no es portador del gen alterado la descenddencia no estara en riesgo de padecer la enfermedad y sus complicaciones, aunque sí podría ser portador de la sicklemia.
Si te interesa conocer sobre la sicklemia aspectos tales como su causa, origen, diagnóstico y tratamiento, continúa leyendo este artículo que te aportará detalles sobre este apasionante tema.
Contenido
¿Qué es la sicklemia y cuál es su causa?
Los avances en el conocimiento de la causa de la sicklemia han servido de modelo para demostrar cómo cambios muy pequeños en los genes que portan la información sobre la estructura de las proteínas, pueden dar lugar a todo un cuadro clínico complejo y mortal. A este tipo de afecciones se les denomina enfermedades moleculares.
La sicklemia, tambien llamada drepanocitosis o anemia drepanocítica es una enfermedad hereditaria, que afecta la estructura de la hemoglobina. Por ello, se clasifica dentro del grupo de las hemoglobinopatías al ser causada por una mutación genética
La causa de la sicklemia radica en una mutación o cambio irreversible en el gen que porta la información genética para la estructura de las cadenas beta de la hemoglobina, proteína que participa en el transporte del oxígeno a los tejidos.
El cambio que se produce en particular es en uno de los bloques o aminoácidos que componen a las cadenas beta de la hemoglobina, pero esto solo, es capaz de dar lugar a una enfermedad que puede llegar a ser fatal. La hemoglobina anormal que se origina se denomina hemoglobina S.
Como la mutación se ubica en la molécula portadora de la herencia, es decir el ácido desoxirribonucleico o ADN, la información anormal sobre la estructura de la hemoglobina se transmite también a la descendencia.
¿Cuáles son las alteraciones funcionales que provoca la hemoglobina S?
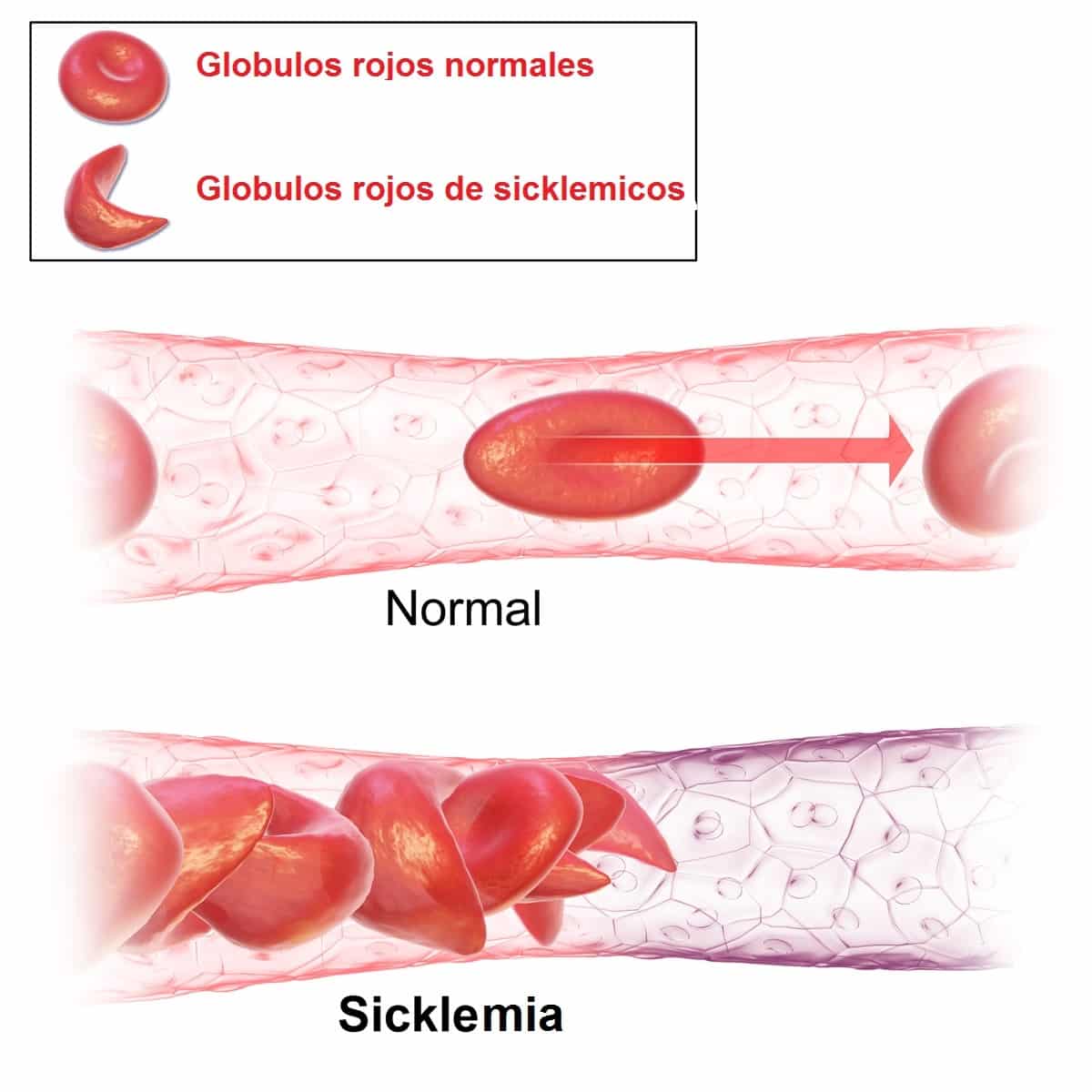
La hemoglobina S, tiene menor afinidad por el oxígeno que la hemoglobina del adulto o hemoglobina A. Esto hace que en condiciones de baja presión parcial de oxígeno la hemoglobina S se polimerice y provoque una deformación de las células sanguíneas que la contienen, que son lo eritrocitos o hematíes. Estas células toman forma de platanito, o luna creciente con extremos alargados y puntiagudos.
Además estos eritrocitos denominados drepanocitos o células falciformes, son más rígidos y frágiles por lo que se rompen con facilidad liberando su contenido, a lo que se denomina hemólisis.
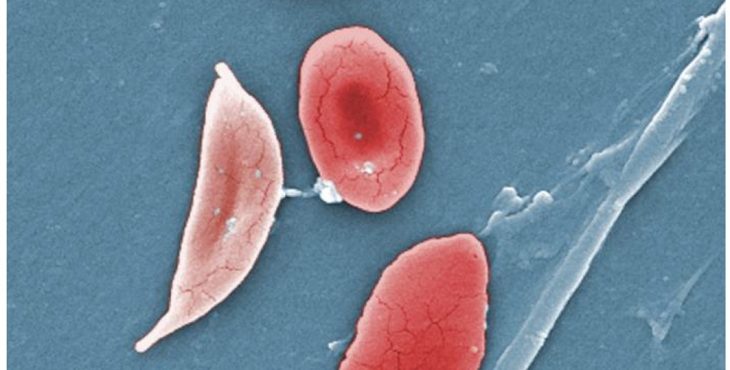
¿Por qué la sicklemia se relaciona con la ascendencia africana?
África desde tiempos remotos, ha sido asolada por una enfermedad terrible denominada malaria o paludismo, que es causa común de muerte en edades tempranas de la vida. Esta enfermedad está causada por un parásito.
Los eritrocitos que contienen hemoglobina S son más resistentes a dicho parásito causal y por ello las personas que portan esta mutación sin padecer la sicklemia, sobreviven más a la malaria, lo que les permite llegar a la edad fértil. Se cree que esto ha proporcionado una ventaja selectiva a estas personas portadoras del gen anormal, y provocado que el mismo se transmita a las generaciones subsiguientes tanto de origen africano como asiatico.
¿Por qué se puede portar el gen de la sicklemia y no experimentar la enfermedad?
Todos poseemos en nuestro ADN un doble juego de genes para codificar la estructura de la hemoglobina. Uno de los juegos proviene de la madre y el otro del padre. Por ello es muy importante determinar la condición de la pareja y así poder evaluar el riesgo en la descendencia.
Para que una persona experimente la sicklemia con su variedad de síntomas y consecuencias, debe poseer ambos juegos de genes anormales. Es decir, que tanto los genes provenientes de la madre como los provenientes del padre, contengan la mutación que origina a la hemoglobina S. Entonces estos individuos son homocigóticos.
Sin embargo, más frecuentemente, solo uno de los padres transmite el gen anormal, por lo que estos individuos se denominan entonces heterocigóticos y muy probablemente, al menos en condiciones normales, no experimentarán ninguno de los síntomas de la sicklemia. A este estado de heterocigóticos se le denomina rasgo drepanocítico.
Sin embargo, como uno de sus juegos de genes está afectado, podrá transmitirlo a su descendencia con una probabilidad que generalmente es de un 50%. Por lo tanto, los heterocigóticos para la sicklemia se consideran portadores de la enfermedad.
¿Qué alteraciones puede sufrir una persona con sicklemia?
Para los homocigóticos la variedad de síntomas y complicaciones es amplia. Tanto es así, que si no reciben tratamiento temprano y sistemático, las personas con sicklemia pueden morir en la niñez o en la primera juventud.
Las alteraciones incluyen:
⦁ Anemia hemolítica. Esto le ha dado también el nombre de anemia drepanocítica a la enfermedad. Implica una disminución de la cantidad de hemoglobina y del conteo de eritrocitos debidos a la ruptura de los mismos o hemólisis ya mencionada.
⦁ Coloración amarillenta de piel y mucosas o ictericia leve. Esta es provocada por el aumento en la degradación de la hemoglobina liberada durante la ruptura de los eritrocitos, que da lugar a la formación de pigmentos amarillos.
⦁ Palidez. Se debe a la anemia o baja concentración de hemoglobina, que es de color rojo intenso.
⦁ Trombosis venosas. Se debe a que el eritrocito que contiene hemoglobina S está deformado y rígido, lo que le impide adaptarse a los vasos sanguíneos estrechos y con frecuencia origina taponaduras o trombosis.
⦁ Infartos. Se refiere a la muerte de los tejidos por falta de oxígeno. Esto se debe a que los eritrocitos en la sicklemia se adhieren a los vasos sanguíneos de pequeño calibre. De esta forma, obstruyen o taponan dichos vasos impidiendo la oxigenación de los tejidos. Pueden afectar a cualquier órgano del cuerpo.
⦁ Trastornos e insuficiencia del desarrollo. El tronco es corto en relación a las extremidades que son largas, mientras el cráneo tiene forma de torre.
⦁ Aumento de tamaño del hígado y del bazo en los niños o hepatoesplenomegalia.
⦁ Aumento de tamaño del corazón o cardiomegalia y soplos.
⦁ Piedras o cálculos biliares. Pueden ocurrir tan tempranamente como a los dos años de edad. Se cree que se deben a la hemólisis. Esta condición es influida por la dieta y otros factores genéticos.
⦁ Úlceras crónicas con forma de sacabocado.
⦁ Crisis dolorosas. Se caracterizan por dolor muy intenso en las articulaciones, en los huesos de las piernas, los pies y las manos. Se deben a la isquemia e infartos provocados por la oclusión de los vasos sanguíneos. También pueden sufrir dolor abdominal.
Para leer mas: el dolor abdominal, ¡que no pase desapercibido!
⦁ Síndrome torácico agudo. Es más frecuente en los niños. Se produce por la obstrucción y cierre de los vasos sanguíneos de muy pequeño calibre. Caracterizado por fiebre, dolor torácico e infiltrados pulmonares. Puede complicarse con una neumonía bacteriana. Frecuentemente conduce a la muerte. Si se produce repetidamente puede dar lugar a hipertensión pulmonar.
⦁ Osteoporosis y disminución de la densidad mineral ósea. Se cree que puede dar lugar a fracturas.
⦁ Expansión de la médula ósea. Se debe a la activación de la formación de eritrocitos a partir de este tejido, con el objetivo de suplantar a los que están resultando destruidos a mayor velocidad de la normal por hemólisis.
⦁ Priapismo. Se produce principalmente en adolescentes jóvenes. Consiste en la erección sostenida del pene de forma anormal no asociada al deseo sexual, y que puede ser causa de disfunción eréctil.
¿Las personas portadoras pueden tener algún síntoma de sicklemia?
Los heterocigóticos para la sicklemia generalmente no presentan síntomas. Sin embargo, esto es en condiciones normales; pero en algunas situaciones especiales, puede variar. De ahi que se recomienden algunas medidas de protección.
Por ejemplo, si la persona se somete a condiciones de baja presión parcial de oxígeno, o hipoxia, como ocurre digamos, en las grandes alturas sobre el nivel del mar, puede sufrir alguna de las complicaciones descritas.
También puede producirse una condición denominada rabdomiolisis, que consiste en la ruptura de las células musculares, particularmente durante el ejercicio muy intenso. Esto puede llevar incluso a la muerte repentina.
Además puede experimentarse una situacion que implica la incapacidad de concentrar la orina o se puede orinar con sangre. Aunque no es frecuente en heterocigóticos, también puede aparecer una alteración del riñón denominada necrosis papilar renal.
¿Cómo se diagnostica la sicklemia?
Existe una prueba de tubo que detecta rápidamente la hemoglobina S basada en su diferente solubilidad respecto a la hemoglobina A. Esta se aplica generalmente en personas que no tienen síntomas pero sí familiares cercanos con sicklemia.
En personas con signos y síntomas se indican pruebas para detectar anemia y su tipo. Se constata así la disminución de la hemoglobina y de los conteos de eritrocitos.
También se aplica una prueba denominada electroforesis de proteína que permite identificar a la hemoglobina S basada en sus diferencias de comportamiento en un campo eléctrico respecto a la hemoglobina normal o hemoglobina A.

Los homocigóticos mostrarán solo o fundamentalmente la hemoglobina S, y pequeñas cantidades de hemoglobina fetal o hemoglobina F. Por otra parte los heterocigóticos mostrarán más cantidad de hemoglobina A que de hemoglobina S. Se indica frecuentemente en estudios prenatales.
Por último resulta necesario observar al microscopio a los drepanocitos o eritrocitos con la deformación característica de la sicklemia para confirmar el diagnóstico.
El diagnóstico prenatal se ha visto favorecido por el advenimiento de modernas técnicas como la reacción en cadena de la polimerasa o PCR.
¿Cuál es el tratamiento de la sicklemia?
Con los adelantos de la ciencia se ha podido lograr que los pacientes con sicklemia superen los 50 años lo cual no era usual con anterioridad.
No existe ningún medicamento que permita curar o prevenir la sicklemia. Por lo tanto, el tratamiento consiste en detectar y tratar a tiempo las diferentes complicaciones.
Se debe hospitalizar a la persona si se sospecha alguna complicación seria como las infecciones graves. Estas infecciones deben ser detectadas y tratadas tempranamente. En los niños se suele utilizar antibióticos de forma preventiva.
En ocasiones las transfusiones de sangre son necesarias.
¿Es posible evitar las crisis y complicaciones de la sicklemia?
Diferentes medidas se dirigen a disminuir el impacto de la sicklemia sobre la calidad de vida de las personas.
Es muy importante el diagnóstico prenatal para poder implantar desde el nacimiento las medidas preventivas.
Se deben utilizar vacunas contra microorganismos productores de infecciones como las meningitis, las neumonías y la gripe. También se indican complementos dietéticos como el ácido fólico y medicamentos como la hidroxiurea. Esta última disminuye la formación de drepanocitos.
Actualmente se considera y estudia la influencia de los factores ambientales en el desarrollo de crisis y complicaciones de la sicklemia. Los factores ambientales que probablemente son importantes incluyen el clima, la calidad del aire, el nivel socioeconómico, los ejercicios y las infecciones.
Al igual que para otras enfermedades genéticas la terapia con células madres es promisoria para lograr la cura de la sicklemia.
Más temas de interés: ¿CONOCES LA IMPORTANCIA DE TOMAR ÁCIDO FÓLICO EN EL EMBARAZO?
